This is a simple initial Python script to calculate the GC content of DNA/RNA sequences, specifically for cases where each FASTA file contains only one sequence.
withopen(infile) as f: f=f.read() f=re.sub('>.*\n|\n', '', f) """Remove the header line and newline characters"""
size=len(f) nG=f.count("g")+f.count("G") nC=f.count("c")+f.count("C") percent_GC=(nG+nC)/size withopen(outfile, 'w') as out: out.write("ID\tsize\tpercent_gc\n") ID=re.sub('.fasta', '', infile) out.write("{}\t{}\t{}\n".format(ID, size, percent_GC)) """Add a newline character (\n) at the end of each line in output_file"""
#use : python3 GC_content.py input.fa out.file
.
After encapsulating the script into an executable file named GC_content.py , then use it on Linux as follows:
1
ls *fa |while read id ;do(python GC_content.py $id ${id}.out.txt);done && cat *out.txt > output.txt
.
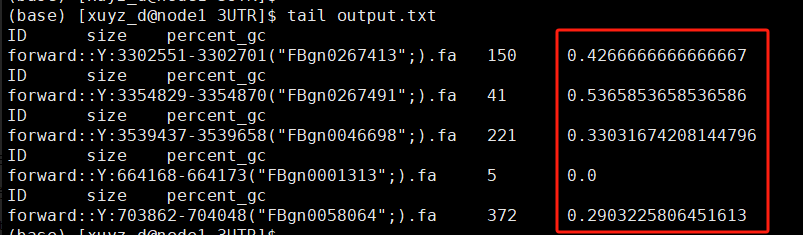
output:
The last column is the calculated GC content ratio for the corresponding sequence.
.
.
If a the fasta file contains multiple sequences, we can use this python script following:
defcalculate_gc_content(sequence): """define a function to calculate the GC percentage for sequence""" sequence = sequence.upper() gc_count = sequence.count('G') + sequence.count('C') total_count = len(sequence) if total_count == 0: return0 return (gc_count / total_count) * 100
defprocess_fasta_file(input_file, output_file): """process a fasta file and calculate the GC content of each sequence""" withopen(output_file, "w") as output: # 读取FASTA文件 withopen(input_file, "r") as file: """file is input_file""" for record in SeqIO.parse(file, "fasta"): seq_id = record.id """Obtain the ID of the current sequence,usually the part following the > in the FASTA file header""" sequence = str(record.seq) """Convert the sequence object (record.seq) into a string format""" gc_content = calculate_gc_content(sequence) output.write(f">{seq_id} GC_content: {gc_content:.2f}%\n") """the format for output.write is: >sequence ID GC_content: GC content%, where .2f indicates that the GC_content will be rounded to two decimal places"""