Transcript factor annotation for target genes .
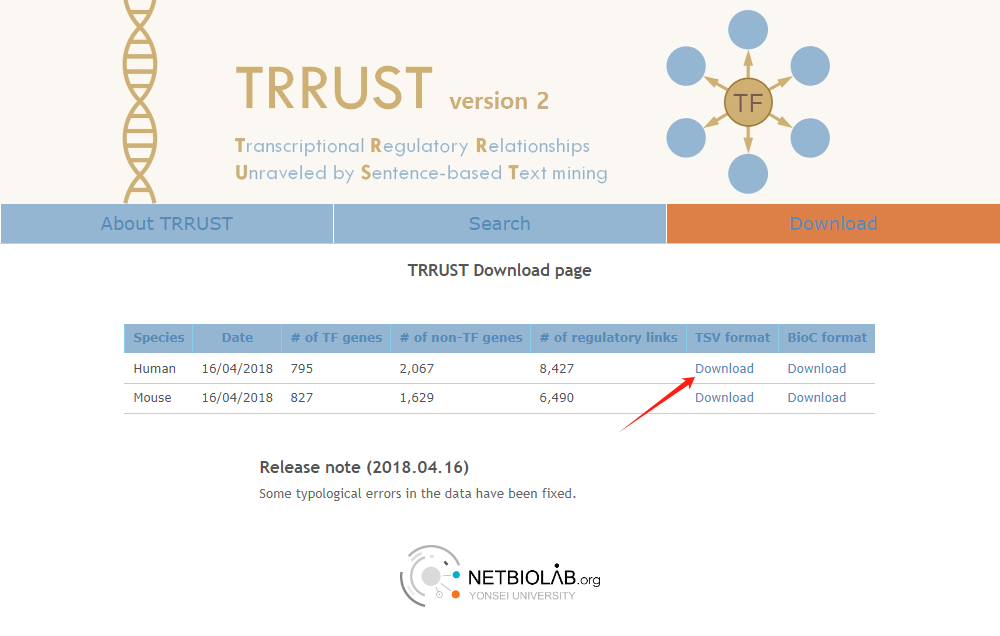
First, I downloaded the data on human transcription factors and their target genes from https://www.grnpedia.org/trrust/downloadnetwork.php , as shown in the picture below.
.
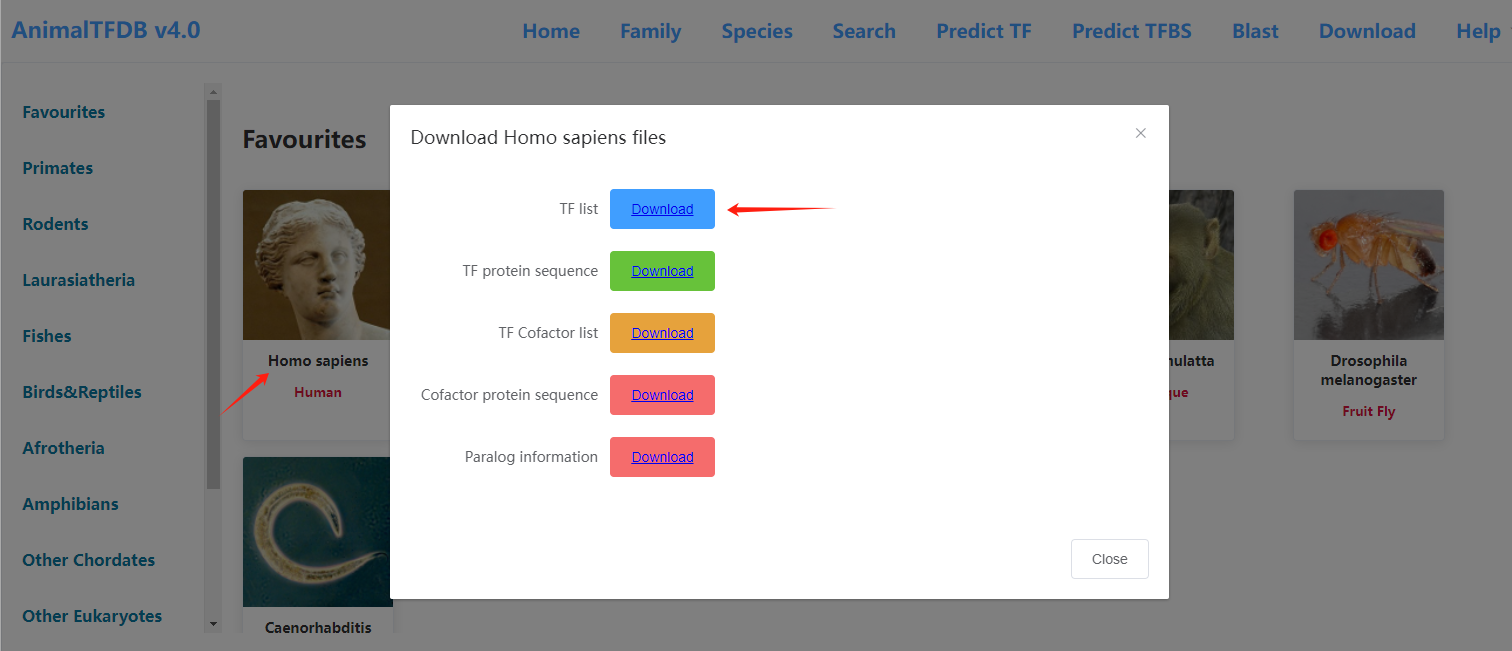
and downloaded the table that lists transcription factors and their corresponding transcription factor families from https://guolab.wchscu.cn/AnimalTFDB4//#/Download , as illustrated below:
.
the pipeline of TF_annotation of DEGs is as follows.
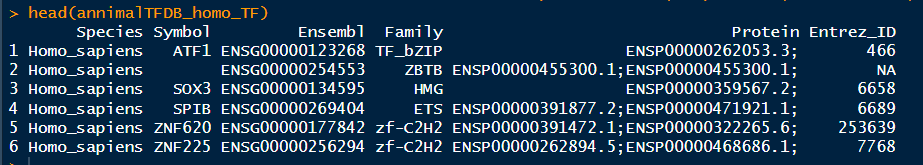
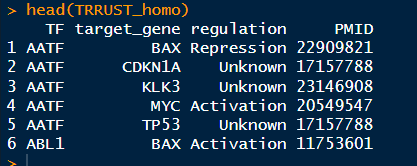
1 2 3 4 5 6 7 8 9 10 11 12 13 14 ( "E:/220625_PC/R workplace/230922_HJ/250305_TF/" ) <- read.table( "animalTFDB_Homo_sapiens_TF.txt" , sep = '\t' , fill = TRUE , header = T ) ( annimalTFDB_homo_TF) <- read.table( "trrust_rawdata.human.tsv" , fill = TRUE , header = F ) ( TRRUST_homo) <- c ( "TF" , "target_gene" , "regulation" , "PMID" ) ( TRRUST_homo) length ( intersect( TRRUST_homo$ TF, annimalTFDB_homo_TF$ Symbol) )
.
load your genes (target genes)
1 2 3 4 5 6 7 8 9 10 11 12 ( "E:/220625_PC/R workplace/230922_HJ/250305_TF/Differentially_expressed_gene/Diff_exp/" ) ( readxl) <- read_xlsx( "df_GFP.VEGF_shSND1.VEGF.xlsx" , sheet = "gene_diff" , skip = 0 ) ( df_GFP.VEGF_shSND1.VEGF) <- c ( 'gene_symbol' , 'log2FC' , 'pvalue' ) $ log2FC <- as.numeric ( df_GFP.VEGF_shSND1.VEGF$ log2FC) $ pvalue <- as.numeric ( df_GFP.VEGF_shSND1.VEGF$ pvalue) <- df_GFP.VEGF_shSND1.VEGF[ abs ( df_GFP.VEGF_shSND1.VEGF$ log2FC) > 1 & $ pvalue < 0.05 , ] length ( intersect( TRRUST_homo$ target_gene, DEG_GFP.VEGF_shSND1.VEGF$ gene_symbol) )
.
merge the table and get the target genes of TF
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 <- TRRUST_homo[ , 1 : 3 ] ( TF_target) <- c ( 'Symbol' , 'gene_symbol' , 'regulate' ) <- merge( TF_target , DEG_GFP.VEGF_shSND1.VEGF, by= "gene_symbol" , all.y = T ) <- na.omit( DEG_tf) length ( unique( DEG_tf$ gene_symbol) ) <- merge( DEG_tf, annimalTFDB_homo_TF, by= 'Symbol' , all.x = T ) <- na.omit( DEG_tf_family) length ( unique( DEG_tf_family$ gene_symbol) ) ( DEG_tf_family$ Family) ( DEG_tf_family, file = 'DEG_tf_family.csv' )